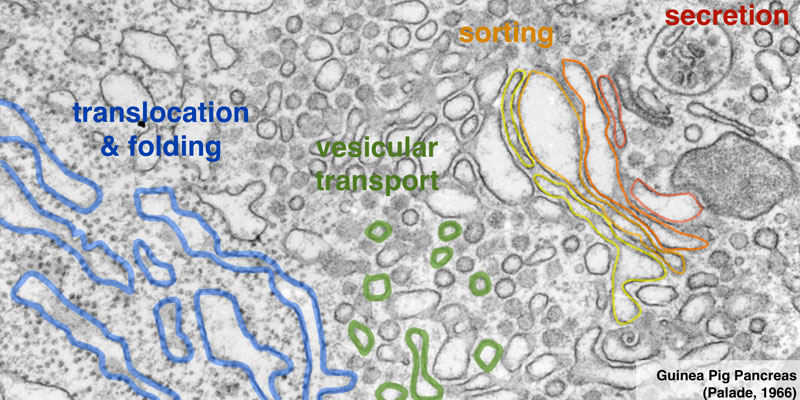
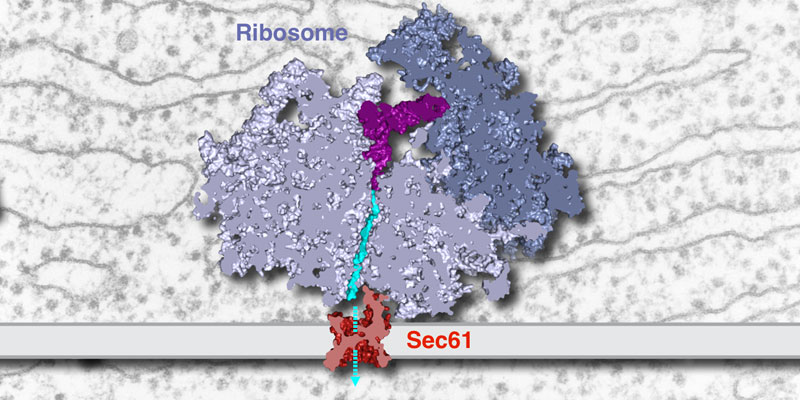
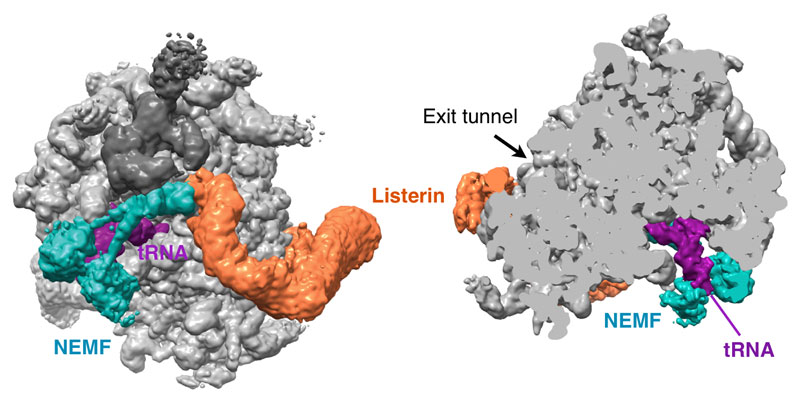
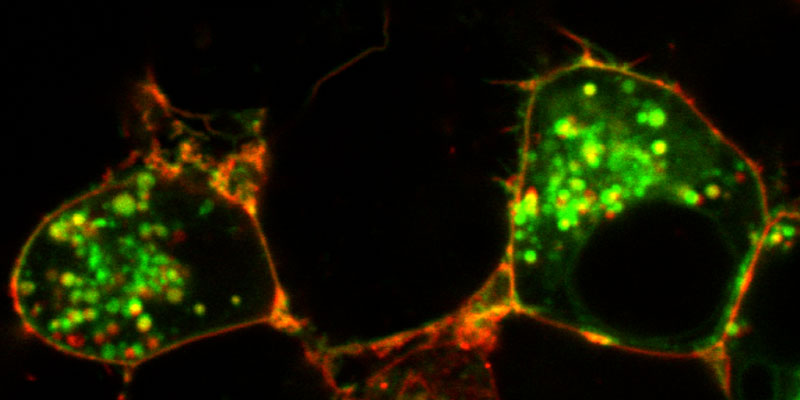
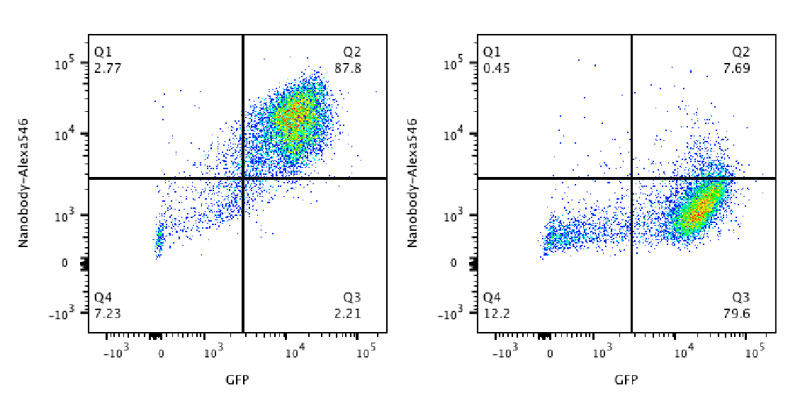
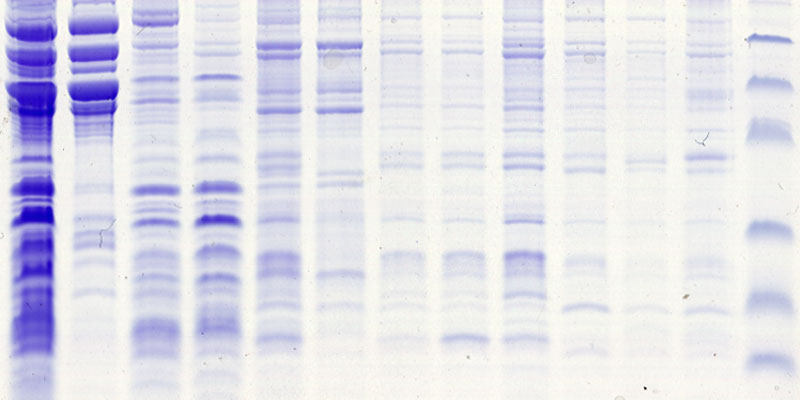
Our research group seeks to answer two related questions. First, how do newly made proteins get to the right part of the cell and assemble into functional products? Second, how do cells recognize errors during protein maturation and target the defective products for degradation? Both processes are essential to all living cells, and even subtle problems in protein maturation or quality control lead to various diseases.