
| BALBES - Automatic Molecular Replacement Pipeline |
Please cite the following paper in any publication arising from use of BALBES
F.Long, A.Vagin, P.Young and G.N.Murshudov
"BALBES: a Molecular Replacement Pipeline " Acta Cryst. D64 125-132(2008)
BALBES Manual
This manual is for BALBES, version 1.1.5, on Linux, Mac OS X. Contact Fei Long for more information. Please also email any comments, questions and suggestions. Thank you.
Contents:
Introduction
BALBES is an automated software suite for structure solutions using protein X-ray crystallographic data. Molecular Replacement(MR) is core scientific method. It is called a MR pipeline because it integrates all components, necessary for finding a solution structure by MR, into one system. The system is automated so that a user needs only to provide a sequence file and a X-ray data file. After that BALBES will perform jobs such as data checking, homologous searching using its internal database to generate a variety of template models, and selecting different algorithms to do MR and refinement on those models and their combinations.
BALBES contains three major parts:
- An specifically tailored internal database for searching homologous models
- Scientific programs, such as MOLREP and REFMAC, for MR and refinement and other jobs
- A manager system written in PYTHON scripts that controls process flow and selects algorithms
Go Back to Contents
Download and Install BALBES
Download BALBES
Currently, BALBES releases two pre-compiled versions that work respectively on:
- Linux
- Mac OS X
The latest versions can be download at MRC-LMB site or YSBL site. It is also included in a CCP4 suite. But the version in CCP4 suite is sometime out-dated.
Install BALBES
Perequisites for BALBES:
You need to install CCP4 suite before installing BALBES
Install BALBES under CCP4 Package (replace a old version with a new one)
- After you download one of gziped tar files, e.g. BALBES_1.1.1_DB_Oct_1_2010_Linux.tar.gz, move it under CCP4 package by, e.g.
mv BALBES_1.1.5_DB_Nov_1_2011_Linux.tar.gz $CCP4/share/
cd $CCP4/share/
- Unpack the BALBES package e.g.
tar zxvf BALBES_1.1.5_DB_Nov_1_2011_Linux.tar.gz
(or another *.tar.gz file you downloaded)
- Close the old xterms and open new ones. Re-start ccp4i and select BALBES. The version of BALBES you are using should be the new one.
Install standalone BALBES
If you want to install a version of BALBES under a directory different from where your ccp4 suite is installed
You need to do the following steps.
- After you download one of gziped tar files, e.g. BALBES_1.1.1_DB_Oct_1_2010_Linux.tar.gz, move it to the directory you want to install BALBES, e.g.
mv BALBES_1.1.5_DB_Nov_1_2011_Linux.tar.gz your_dir
cd your_dir
- Unpack the BALBES package you just moved e.g.
tar zxvf BALBES_1.1.1_DB_Oct_1_2010_Linux.tar.gz
cd BALBES
- You will see files README.SETUP and setup.py.
./setup.py or python ./setup.py
The installation script will do everything for you
Go Back to Contents
Update BALBES Database
We update and release the internal database of BALBES for homologous searching every month to include entries related to new structures released by Protein Data Bank(PDB). Presume you have installed BALBES, and would like to use the newly released database of BALBES, you need to do the following steps.
- Download a version of BALBES database, e.g. BALBES__DB.tar.gz.
- Move the database you have downloaded to the directory where your environment variable $BALBES_ROOT defines ($BALBES_ROOT exists if you have installed CCP4 suite).
mv BALBES_DB.tar.gz $BALBES_ROOT
- Unpack the BALBES database you have just moved e.g.
tar zxvf BALBES_DB.tar.gz
After finishing the above steps, BALBES will use the new internal database.
Go Back to Contents
Run BALBES
Because BALBES is designed to minimize user's intervention, run BALBES is very simple. Three ways are provided to run BALBES,
i.e.
- Run BALBES via BALBES webserver
- Run BALBES via CCP4i interface
- Run BBALBES from command-lines
In this section, we will also describe
Requirements for input files
The requirements depend on what you want to do with BALBES. If you would like to
- solve structures using Molecular Replacement, you need to provide two files, i.e.
- a structural factor file of MTZ
or CIF
format, such as mySF.mtz or mySF.cif
- a sequence file in form of FASTA
format, e.g. mySeq.seq
- just search for homologous models, you need to provide one file, i.e.
- a sequence file in form of FASTA
format, e.g. mySeq.seq
Go Back to "Run BALBES"
Run BALBES via BALBES webserver
Run BALBES for structural solutions via BALBES web server
is shown is the following picture:

|
- Upload a structural factor file and a sequence file
- Click submit button
|
Go Back to "Run BALBES"
Run BALBES via CCP4i
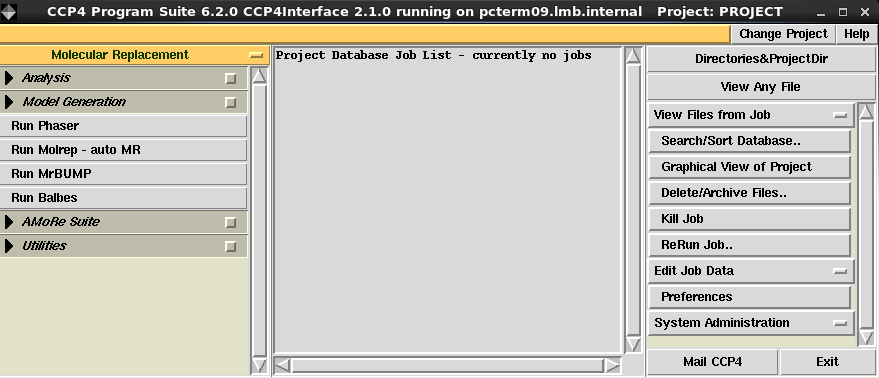
Steps in run BALBES for structural solutions via CCP4i interface are shown in the following pictures.
|
- Select "Molecular Replacement" Module in CCP4i main manu
- Select "Run BALBES" task in Molecular Replacement Module
|
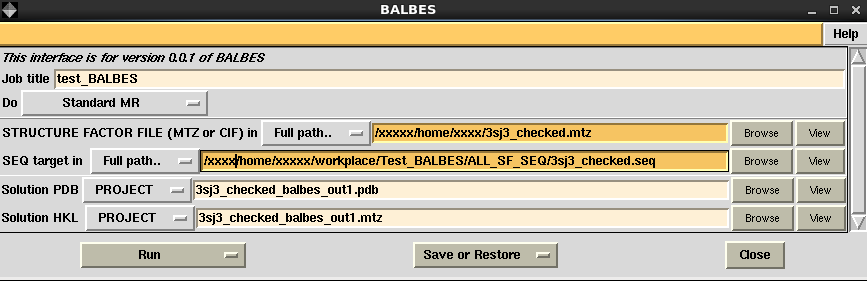
|
- Upload a structural factor file and a sequence file to the input file rows
- Give names to the output PDB and MTZ files, otherwise default names will be applied
- Click "Run" button
- Other steps (such as view the results) are standard procedures
in ccp4i. Please consult with ccp4i's help manual if you are not familiar
with them.
|
Go Back to "Run BALBES"
Run BALBES from command lines
Typical command line for structural solution is :
balbes -o a_directory_name -f
a_structure_factor_file_name -s a_sequene_file_name
Explain:
- program name: balbes
- options (include symbol - ):
-o (optional) ------- following by the name of
the directory into which you want to put all of the output
generated by balbes.
e.g. -o MyjobOne
or -o ../output_abcd
If the directory does not exist, the program
will create the directory( MyjobOne,
or ../output_abcd) for you.
If -o is omitted, the program will
create a output directory called
output_username (e.g. output_fei)
- -f (required) ------- followed by the name of a structural factor file.
The file could be a .mtz file or a .cif file.
If the file is in the current directory, you can just type name, e.g,
-f 1ab2.mtz
or -f 3cd4.cif
Otherwise you need to input the file name
with the absolute path. e.g,
-f /home/dir1/dir2/1ab2.mtz
or -f ../3cd4.mtz
- -s (optional) -------- followed the name of a sequence
file. You can find the format of a sequence files containing
one sequence
or multiple sequences
Depending on where the file is,it can be,
-s aaaaa.seq
or -s ../../aaaa.seq
or -s /Users/abcd/data/aaaa.seq
Go Back to "Run BALBES"
All BALBES command line arguments:
- -o
- Root directory for BALBES job output. It contains following subdirectories:
results -- stores the final solutions in terms
of pdb and mtz files, and a file recording over-all process
process_details -- stores all
*.log, *.ps files, generated by individual programs within BALBES
during executing balbes.
template_models--contains all homologous models found
- -f
- Input x-ray data file of MTZ or CIF format
- -s
- Input sequence file of FASTA format
- -m
-
Input model file of PDB format. BALBES will no long search for homologous models, just use the provided one
It can be,
-m aaaaa.pdb
-m ../../aaaa.pdb
-s /Users/abcd/data/aaaa.pdb
- -l
-
user's own database. BALBES will search homologous models using that database instead of its internal
database. The user's own database is actually a subdirectory contains a group of user's model files
of PDB format.
For example, the user's database is called myLib, then
a_user$ ls myLib/
a_user$ 1.pdb 2.pdb 12.pdb 26.pdb aaaa.pdb xxxx.pdb yyyy.pdb
a_user$ balbes -o myJobOut -f myMTZ.mtz -s mySeq.seq -l myLib/
Go Back to "Run BALBES"
Go Back to Contents
Basic Tutorials