Neurodegenerative diseases are characterised by the pathological assembly of specific proteins into filaments within the central nervous system. Genetic studies have established a causal role for assembly in neurodegeneration. Our goal is to understand the molecular mechanisms of pathological protein assembly in motor neuron diseases (including amyotrophic lateral sclerosis) and frontotemporal dementias, two related groups of neurodegenerative diseases. We anticipate that this work will reveal strategies to target assembly for the diagnosis and treatment of these devastating diseases.
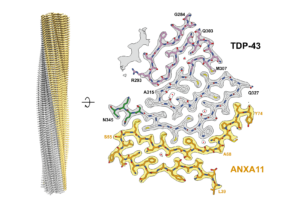
We determine the structures of pathological protein assemblies in neurodegenerative diseases using electron cryomicroscopy (cryo-EM). We have shown that TAR DNA-binding protein 43 (TDP-43) forms distinct amyloid filament structures in different diseases. We have discovered previously unknown pathological assemblies, including amyloid filaments of TATA-binding protein-associated factor 15 (TAF15) and heteromeric filaments of TDP-43 and annexin A11. These findings have transformed our understanding of the molecular basis of several neurodegenerative diseases and introduced new targets for diagnosis and therapy.
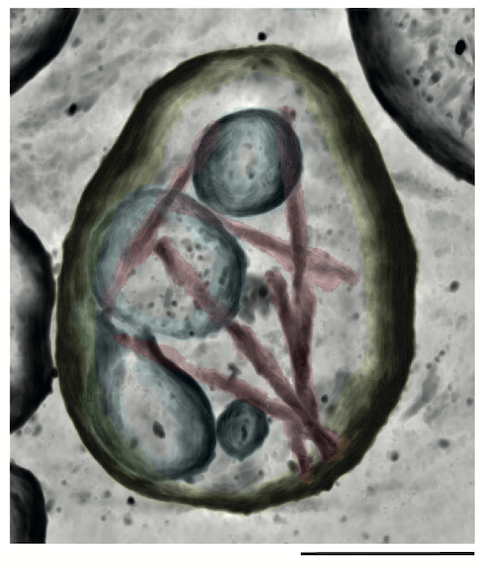
We study pathological protein assembly in its cellular and tissue contexts using molecular-resolution electron cryotomography (cryo-ET) and targeted proteomics. This has revealed unexpected molecular environments and interactions that may play important roles in assembly and neurodegeneration. We are using the knowledge gained through these studies to develop model systems that recapitulate disease-characteristic filament structures. Such models are required to further investigate the molecular mechanisms of neurodegenerative diseases.