Mutagenesis and DNA damage tolerance
Cellular DNA requires continuous maintenance to counter the effects of spontaneous degradation and exogenous damage. All forms of life possess robust repair mechanisms that can excise damaged bases since these will interfere with essential cellular processes, especially transcription and replication. DNA damage encountered during replication poses a particular problem. The replicative polymerases are highly accurate and one reason for this is that they do not tolerate damaged bases on the DNA template. A damaged template leads them to stall and thus blocks progression of the replication fork.
Attempts to excise the DNA damage at the replication fork will result the formation of a double strand break. The cell can attempt to use homologous recombination to repair the break and restart of the fork. Failure results in genetic instability or cell death. To avoid this the replication machinery attempts to bypass the damage, deferring definitive repair of the lesion until later when a stable duplex is re-established. Cells have has two basic strategies for bypassing, or tolerating, DNA damage:
1. Template switching
In template switching, the stalled polymerase finds an alternative template that the the stalled replicative polymerases can 'borrow' to get around the lesion and restart replication. This is most commonly the newly synthesised daughter strand on the sister chromatid.
2. Translesion synthesis
Alternatively, the cell can switch polymerases replacing the replicative polymerases with a combination of specialised translesion polymerases that are able to synthesise directly across the lesion. This process is known as translesion synthesis (TLS). This mode of bypass carries a significant risk of introducing a mutation as the template base is non- or mis-instructional, and the TLS polymerases are intrinsically error prone.
Error-prone lesion bypass can make sense in a single celled organism. In a changing environment becoming a mutator can be beneficial, at least to the population as a whole. Such behaviour is seen in the SOS response in E. coli, in which stress leads to increased activity of translesion polymerases. However, similar behaviour in the somatic cells of multicellular organisms could result in loss of the normal control of cell growth and division and the development of tumours. It was therefore rather a surprise when, in the late 1990s, it became clear that vertebrates possessed homologs of the bacterial and yeast translesion polymerases. Further, we and others have shown that these enzymes play a critical role in the vertebrate DNA damage response.
Our work in this area has focussed on the mechanisms by which the translesion polymerases are controlled, and how translesion synthesis interacts with recombinational modes of DNA damage bypass. A particularly important finding for us was the identification of specific genetic requirements for translesion synthesis at and behind the replication fork.
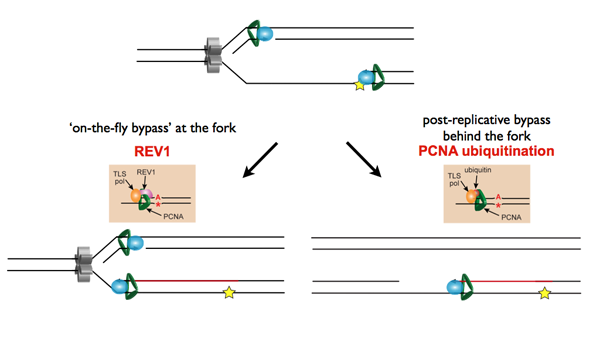
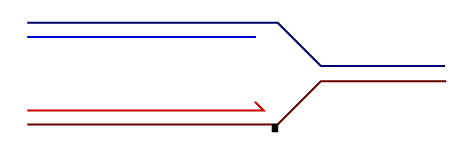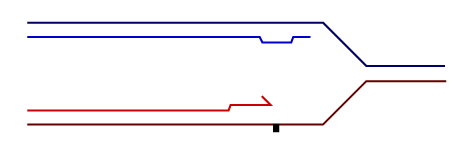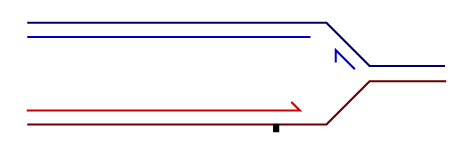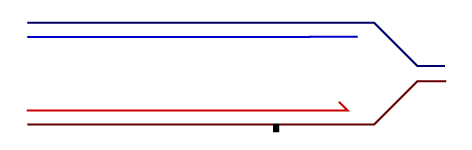
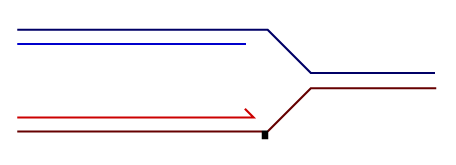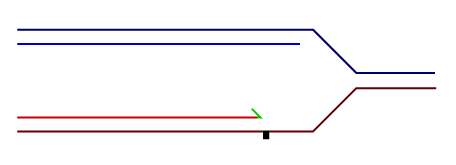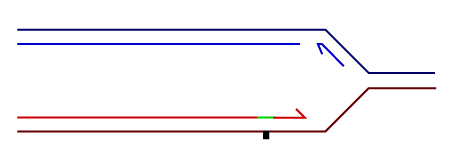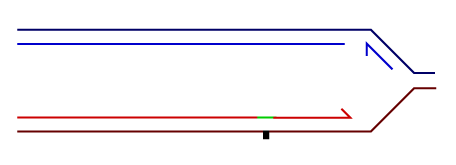
Temporally distinct modes of DNA damage bypass
A replication fork with leading strand synthesis blocked by a DNA lesion. Replication can continue either by initiating bypass of the lesion directly allowing the blocked leading strand polymerase to continue. We have termed this 'on-the-fly' bypass and have shown that it requires the TLS coordinating function of REV1. Alternatively, replication can reprime downstream of the lesion leaving a single stranded gap. The lesion is then bypassed and the gap filled later on. This delayed mode of lesion bypass is dependent on PCNA ubiquitination.
It was this observation that prompted us to start thinking about the effects the timing of lesion bypass have on histone management during replication and on the maintenance of epigenetic memory.
Our work on the impact of replication impediments on histone management also led us to explore the role of the variant histone H3.3 in the DNA damage response and in mutagenesis. H3.3 differs from the canonical H3 by only four amino acids. However, this small difference confers very distinct properties on nucleosomes containing this histone. We have uncovered an unanticipated role for H3.3 during replication of UV-damaged DNA and in cellular resistance to UV irradiation and other forms of DNA damage. We have also discovered that H3.3 influences the formation of single stranded DNA during transcription of the immunoglobulin variable genes. In turn, this influences the ability of activation induced deaminase, the enzyme that drives immunoglobulin diversification, to mutagenise the DNA of the IgV genes.
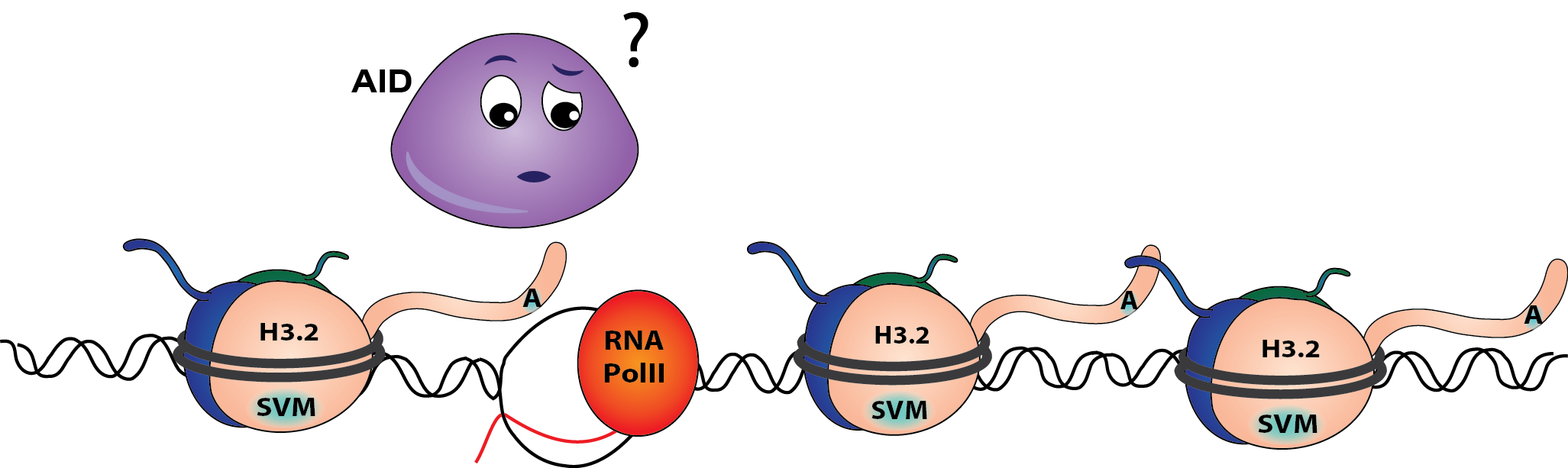
H3.3 promotes effective immunoglobulin mutagenesis
In a DT40 cell line lacking H3.3 completely, AID struggles to access the IgV genes resulting in poor mutagenesis. This is because less single stranded DNA is exposed during IgV transcription. Interestingly, the efficiency and kinetics of transcription are not affected.
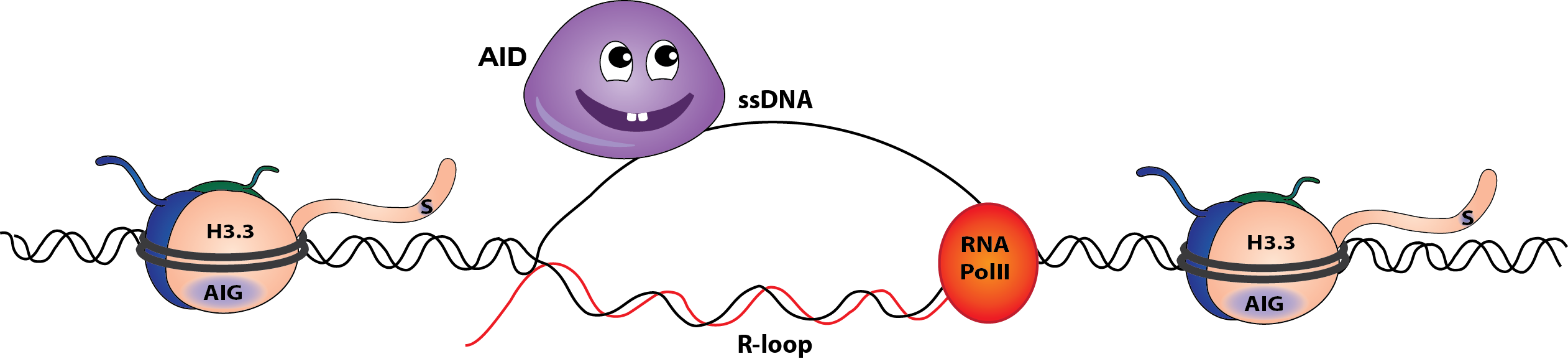
When H3.3 is present, the formation of single stranded DNA and AID efficient AID-dependent Ig mutagenesis is enhanced. (Cartoons by Marina Romanello)
We are continuing to explore the mechanisms by which H3.3 influences these processes and the extent to which the mutations in H3.3 that have been described in a range of paediatric tumours could contribute to genetic instability.