e have developed ‘photochemical genetic’ methods to rapidly control the activity of proteins in living cells, providing insight into the dynamics of elementary steps in biological processes, as well as insight into the regulation of intracellular network connectivity in space and time; with these methods, we hope to understand molecular processes inside cells and organisms with the level of precision more commonly associated with in vitro biochemistry or biophysics.
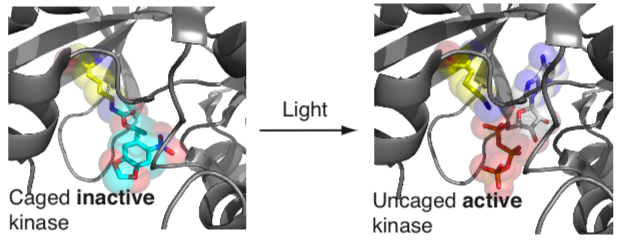
To directly and specifically control molecular functions of user-defined proteins, with high spatial and temporal precision, in living cells and organisms we have developed approaches for site specifically incorporating photocaged versions of natural amino acids (including lysine, tyrosine and cysteine) into proteins. We have demonstrated that protein interactions, protein translocation, protein catalytic activity, proteolysis and the post-translational modification of proteins can be regulated by replacing crucial natural amino acids in proteins with their genetically encoded photocaged version. Using this approach, an initially inactive protein, in which a key residue is photocaged, can be rapidly activated by shining a millisecond pulse of light on cells to remove the photocage and reveal the active protein.
Protein trafficking. In early experiments we demonstrated that photocaging a key lysine residue in a nuclear localization sequence allowed mis-localization of a fluorescently tagged protein to the cytosol of mammalian cells. Upon illuminating the cells with a brief pulse of light we triggered translocation of the protein to the nucleus and followed the kinetics of this process in single cells by fluorescence microscopy. These experiments allowed us to measure the kinetics of nuclear transport, and provide a general route to rapidly re-localize a protein to control its function. In additional experiments we were able to cage a key lysine in a nuclear localization signal in p53, and define the contribution of that signal to the kinetics of nuclear import.
Movie 1. Photocontrol of a caged NLS, at time 0 in the movie, followed by fluorescence microscopy.
Insights into kinase signalling networks, enzyme caging and substrate caging. Cellular signalling transmits information about the environment to the cell through a network of protein phosphorylation events. A challenge in understanding cell signalling is to understand this information transmission in space and time. What are the kinetics of elementary steps in signalling? how do adaptive feedback mechanisms control signalling in space and time? How do we design inhibitors that target the correct part of the network? These questions have been very challenging to address by classical genetics and SiRNA approaches because the networks are sufficiently plastic that they often adapt on the time scale of the perturbation. Ideally we could rapidly activate any kinase in a signalling pathway and turn on a subnetwork of signalling to dissect the contributions of elementary steps to signalling and elucidate the topology of feedback/feedforward regulation. We have begun to implement a strategy to achieve this, with an initial focus on MAP kinase signalling.
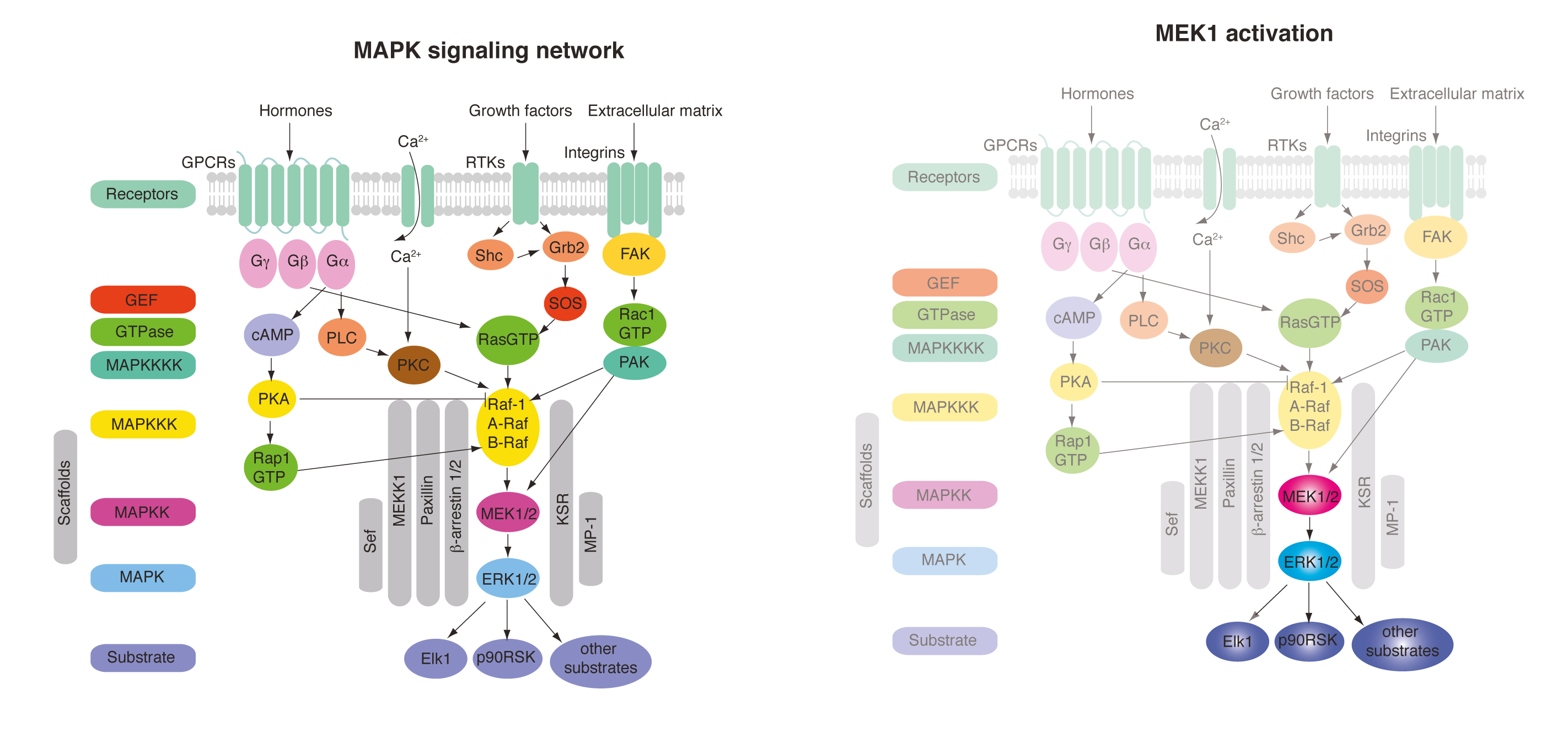
The mitogen-activated protein kinase (MAPK) pathway is a signaling pathway involved in fundamental cellular processes including growth and proliferation. This is typically activated at the cell surface by binding of growth factors, such as epidermal growth factor (EGF) to an epidermal growth factor receptor (EGFR). EGF stimulation leads to relocalization of the downstream factor ERK2 from the cytoplasm to the nucleus, and this can be followed by fluorescence microscopy using an ERK2–GFP fusion. Following EGF stimulation, there is a lag phase before ERK2–GFP accumulates in the nucleus, and ERK2–GFP then dissipates from the nucleus despite persistent EGF stimulation; this latter phenomenon – described as exact adaptation – suggests there may be negative feedback in the pathway.
MEK1 is a kinase which is an integral part of the MAPK signaling pathway and is able to directly phosphorylate ERK2. By introducing mutations to make MEK constitutively active and simultaneously replacing the key lysine residue in the active site of MEK with a photocaged lysine, a photoactivatable version of MEK was created in mammalian cells. When cells were subjected to a pulse of light, active MEK was produced that phosphorylated ERK2–GFP, leading to its nuclear import. The kinetics of this process were followed by fluorescence microscopy, and additional experiments demonstrated that activation of MEK leads to phosphorylation of ERK2, as well as to the phosphorylation of nuclear targets of ERK2. This demonstrated that a subnetwork of MAPK signalling can been specifically photoactivated by a process that is independent of receptor stimulation.
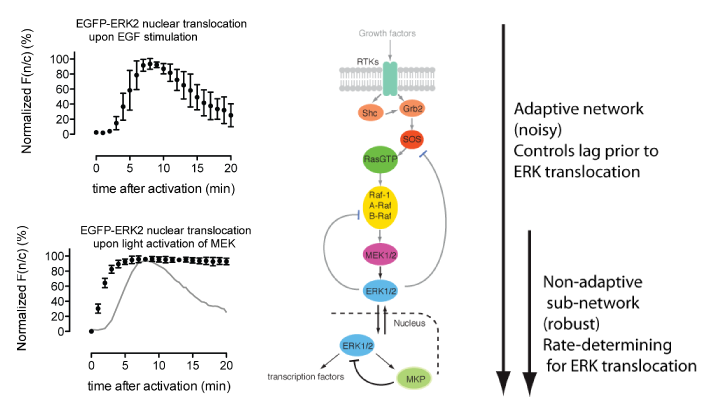
Comparison of the kinetics of ERK2–GFP nuclear accumulation following MEK photoactivation or endogenous receptor stimulation provided some interesting insights. Whereas there is a lag phase of minutes before the nuclear accumulation of ERK2–GFP following receptor stimulation, this long lag phase is not present in the photoactivated subnetwork, suggesting that the lag may result from steps upstream of MEK. Once initiated, the rates of ERK2–GFP nuclear accumulation are comparable, suggesting that the rate-determining step for ERK2 nuclear import may lie between MEK and ERK2. In addition, although exact adaptation is observed in the receptor-stimulated case, this is not observed in the photoactivated subnetwork. This suggests that the feedback mechanism that controls exact adaptation may operate upstream of MEK in the MAPK pathway. Finally, following the import of ERK2–GFP with high time resolution showed that it accumulated sigmoidally, which is consistent with its distributive dual phosphorylation, previously observed in vitro, being rate-determining for nuclear import.
Movie 2. ERK2 translocation kinetics
This approach may also be used to profile the network targets of inhibitors: we showed that a known MEK inhibitor could selectively interfere with the MEK-ERK subnetwork, localizing the activity of the inhibitor to this part of the network.
Movie 3. Inhibitor specificity defined by sub-network profiling. Cells were illuminated at time t = 0 min and U0126 (10 μM) was added after 8 min. The EGFP fluorescence resulting from ERK-EGFP of a representative cell is observed following MEK photoactivation. The graph on the right shows normalized F(n/c) vs. time (mean of 10 independent experiments).
Because the lysine caged in these experiments is almost universally conserved in protein kinases, it should be possible to apply extensions of this approach to provide further insights into the elementary steps in MAP kinase signalling and many other kinase pathways.
In recent experiments we have demonstrated the incorporation of photocage tyrosine into proteins expressed in mammalian cells and caged a tyrosine that is a substrate of a protein kinase. This has allowed us to photocontrol kinase signalling at the substrate level, and this approach should provide complementary information to that obtained by caging kinases.
We have also shown that cysteine residues in cellular proteins may be caged, and have leveraged a genetically encoded photocaged cysteine to create a version of TEV protease that can be activated upon illumination. By introducing TEV sites into proteins of interest it is now possible to optically trigger the cleavage of a target protein.
Publications
Genetic Encoding of Photocaged Cysteine Allows Photoactivation of TEV Protease in Live Mammalian Cells.
J. Am. Chem. Soc. 136, 2240-2243, doi:10.1021/ja412191m (2014).
D.P. Nguyen, M. Mahesh, S. J. Elsaesser, S. M. Hancock, C. Uttamapinant, J. W. Chin.
Genetically encoded light activated transcription for spatiotemporal control of gene expression and gene silencing in mammalian cells
J. Am. Chem. Soc. 2013 135:13433-13439.
J. Hempill, C. Chou, J.W. Chin, A. Deiters.
Photocontrol of Tyrosine Phosphorylation in Mammalian Cells via Genetic Encoding of Photocaged Tyrosine.
J. Am. Chem. Soc. 134, 11912-11915, doi:10.1021/ja3046958 (2012).
E. Arbeley, J. Torres-Kolbus, A. Deiters, J.W. Chin.
Light Activated Kinases Enable Temporal Dissection of Signaling Networks in Living Cells.
J. Am. Chem. Soc. 133, 2124-2127, doi:10.1021/ja1109979 (2011).
A. Gautier, A. Deiters, & J.W. Chin.
[Featured in Nature Meths. 2011 4:284-285, Chem. Eng. News 14 Feb. 2011]
Genetically Encoded Photocontrol of Protein Localization in Mammalian Cells.
J. Am. Chem. Soc. 132, 4086-4088, doi:10.1021/ja910688s (2010).
A Gautier, D.P. Nguyen, H. Lusic, W. An, A. Deiters, J.W. Chin.
[Featured in Chem. Bio Chem. 2010 11:1825-1827]