
 Work published in the journal Structure by Kiyoshi Nagai’s group in the LMB’s Structural Studies Division, has provided further detailed information on the structure and role of proteins at the active site of the spliceosome, and may also help to explain the molecular pathology of the eye disease, retinitis pigmentosa type 13 (RP13).
Work published in the journal Structure by Kiyoshi Nagai’s group in the LMB’s Structural Studies Division, has provided further detailed information on the structure and role of proteins at the active site of the spliceosome, and may also help to explain the molecular pathology of the eye disease, retinitis pigmentosa type 13 (RP13).
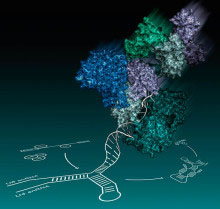
The spliceosome is a dynamic molecular machine responsible for the excision of introns from pre-messengerRNA (pre-mRNA) precursors and ligation of exons to form continuous protein coding sequences in the nucleus. Five RNA-protein subunits, namely U1, U2, U4, U5 and U6 small nuclear ribonucleoprotein particles (snRNPs), and various non-snRNP proteins, assemble on pre-mRNA substrates in a highly ordered manner. The spliceosome then undergoes major compositional and structural changes leading to the formation of the catalytic core consisting of a network of U2, U5 and U6 snRNAs and pre-mRNA. Brr2 helicase, a U5 snRNP protein, unwinds U6 snRNA from U4 snRNA and allows U6 snRNA to engage in an intricate RNA network at the catalytic centre of the spliceosome within Prp8, another U5 snRNP protein. Both Brr2 and Prp8 crosslink with catalytic RNA components and the pre-mRNA substrate and thus they form the active site cavity of the spliceosome.
In work led by Kelly Nguyen, a student in the group, the crystal structure of yeast Brr2 helicase in complex with the Jab1/MPN domain of Prp8 was solved. Brr2 helicase contains a catalytically active N-terminal cassette and an inactive C-terminal cassette. Contrary to previous reports, the crystal structure and mutagenesis data show that the Jab1/MPN domain binds exclusively to the N-terminal helicase cassette of Brr2. Together with the structure of Prp8 published earlier this year, this suggests how the U6 snRNA unwound from U4 snRNA is introduced into the active site of the spliceosome.
The knowledge of Prp8 protein structure, particularly in association with its partners, provides insights into the splicing reaction. The residues in the Jab1/MPN domain, whose mutations in human Prp8 cause the degenerative eye disease retinitis pigmentosa (RP), are found at or near the interface with Brr2. The disease may be caused by a splicing defect of some mRNAs when the Prp8-Brr2 interaction is disrupted. Understanding the molecular pathology of RP13, a genetic disease which leads to blindness, is an important first step towards prevention or delaying the onset of the disease.
This work was supported by the Medical Research Council and a Herchel Smith Research Studentship for Kelly.