
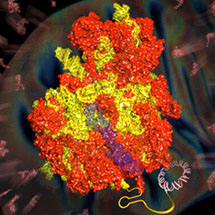
 The bringing together of several independent lines of research by members of the LMB’s Structural Studies Division has led to the determination of the atomic structure of the yeast mitochondrial ribosome. The work took advantage of recent developments in electron microscopy (EM), brought about by a decade of work at the LMB to develop better EM detectors. In a parallel effort, the work also used improvements in the algorithms for computer-based analysis of the “noisy” EM images of single particles. This is the first time that the atomic structure of a large asymmetric complex has been obtained without crystallization. It represents a major scientific breakthrough, that would have been unthinkable just a couple of years ago.
The bringing together of several independent lines of research by members of the LMB’s Structural Studies Division has led to the determination of the atomic structure of the yeast mitochondrial ribosome. The work took advantage of recent developments in electron microscopy (EM), brought about by a decade of work at the LMB to develop better EM detectors. In a parallel effort, the work also used improvements in the algorithms for computer-based analysis of the “noisy” EM images of single particles. This is the first time that the atomic structure of a large asymmetric complex has been obtained without crystallization. It represents a major scientific breakthrough, that would have been unthinkable just a couple of years ago.
Ribosomes are the large assemblies that translate the information in our genes into the thousands of proteins that carry out the many functions in each cell. Mitochondrial ribosomes (mitoribosomes) are indispensable for life as they synthesise essential proteins within the mitochondria. Mitochondria are organelles present in all eukaryotic cells that are mainly responsible for the production of energy. They are thought to be descended from bacteria that were swallowed by a host cell and settled into a mutually beneficial relationship. Although most of the genes that originally resided in the parent of today’s mitochondria are now encoded in the host cell’s nucleus, mitochondria still retain their own small genome. This genome contains codes for only a few proteins, which are made by the mitoribosomes. However, because they are so difficult to purify in large amounts, there has, until now, been no structure of a mitoribosome.
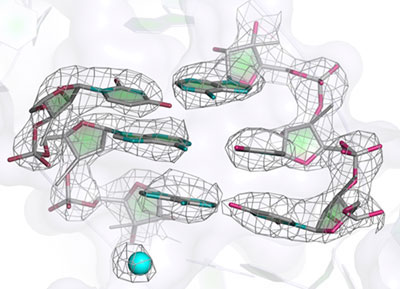
Many genetic diseases are caused by defects in mitochondrial components including their ribosomes. The mitoribosome structure solved, published in the journal Science, reveals a wealth of information. Perhaps most interesting is that the tunnel, through which newly made proteins emerge from the ribosome, takes on a very different path in the yeast mitochondrial ribosome compared to all other ribosomes previously known. Also, many antibiotics that target bacterial ribosomes have toxic side effects because they also affect mitochondrial ribosomes. The structure also hints at why some antibiotics do not bind to mitochondrial ribosomes and are thus relatively non-toxic. This work is just the beginning in our understanding of how mitochondrial ribosomes work and of the differences compared with human cytoplasmic ribosomes, which may have implications for treating disease.
This advance also demonstrates how groups with diverse interests and expertise at the LMB have worked for over a decade, first separately and then in collaboration, to produce a breakthrough in structural biology. Many of the most important processes in cells are carried out by large complexes that are difficult to crystallise for structure determination by traditional methods. Therefore, the methods developed by the different groups involved in this work will also be useful to determine further structures by single particle EM.
Further References:
Paper in Science
Science: Perspective – The Resolution Revolution. Werner Kühlbrandt.Chemical & Engineering News – Electron Microscopy Milestone
Venki’s group page
Richard’s group page
Sjors’ group page
Garib’s group page
Background:
The project was initiated as a result of a chance dinner conversation between John Walker and Venki Ramakrishnan. John, at the time the Director of the MRC’s Mitochondrial Biology Unit, shared the 1997 Nobel Prize for Chemistry for his work on the structure of the enzyme from mitochondria that makes ATP, the energy currency of the cell, namely F1 ATP synthase. Venki shared the 2009 Nobel Prize for Chemistry for his work on ribosome structure and function. Together, they recruited Alexey Amunts from Israel to work on trying to crystallize mitochondrial ribosomes.
After two years of work, it was clear that this was going to be very difficult and the project was almost abandoned. Fortunately, developments in EM detector technology at the LMB, in collaboration with the detector development group at Rutherford-Appleton Laboratory near Harwell, completely changed the state of affairs. Until recently, when applied to randomly oriented particles “frozen” in a thin layer of ice at liquid nitrogen temperatures, the technique could only be used to get low resolution structures which were not detailed enough to deduce an atomic model. For over a decade, Wasi Faruqi and Richard Henderson spearheaded the development of better detectors for EM because a seminal paper in 1995 by Henderson led to his belief that better detectors were essential for getting to high resolution. This led to the development of the prototype of the Falcon II detector in a collaboration with the Max Planck Institutes in Frankfurt and Martinsried and the Rutherford Appleton laboratory, and resulted in its commercialisation by FEI. Apart from being a better detector, the Falcon II is also very fast. Greg McMullen in Richard’s group was able to extract individual frames of the detector so that one could obtain snapshots of the particles while they moved during exposure in the electron beam.
In a parallel effort, Sjors Scheres was leading efforts to determine the best possible way to analyse the very noisy images of single particles in EM images, whose alignment (or orientation) has to be determined accurately to be combined to produce a 3D structure. His program RELION is now used worldwide and is able to provide not only the best possible alignment, but can also optimally separate mixtures of particles present in a sample. Recently his group, working with Israel Sanchez in Venki’s group, had shown that it was possible to get near atomic structures of the ribosome from very small numbers of particles by using the better detector and taking into account the movement of the particles that would normally lead to blurring.
This technical advance persuaded Alexey and Xiaochen Bai (a postdoctoral fellow in Sjors’s group) to collect data on the mitochondrial ribosome from yeast. With careful processing, they were able to obtain a resolution of almost 3 Å, which was previously only possible by x-ray crystallography. This meant that they could see a detailed structure, but interpreting it in atomic terms was a huge undertaking that took months of work, mainly by Alan Brown and Jose Llácer in Venki’s group, with some help from Tanweer Hussain. The problem was that not only were many of the structures of the over 50 proteins in the complex not known, but even the locations of the individual proteins were not known. This required many computational tools to be brought together. Paul Emsley, the author of the most widely used model-building program Coot, was able to modify it to quickly build new proteins into the EM maps and optimise their fit. When structures are initially placed into maps, they are crude and are not chemically correct in their geometry. The final chemically more accurate structures are a result of refinement. Here again, it was fortunate that Garib Murshudov and his associate, Fei Long, also in the Structural Studies Division were able to modify Garib’s widely used refinement program REFMAC for use with EM maps.