
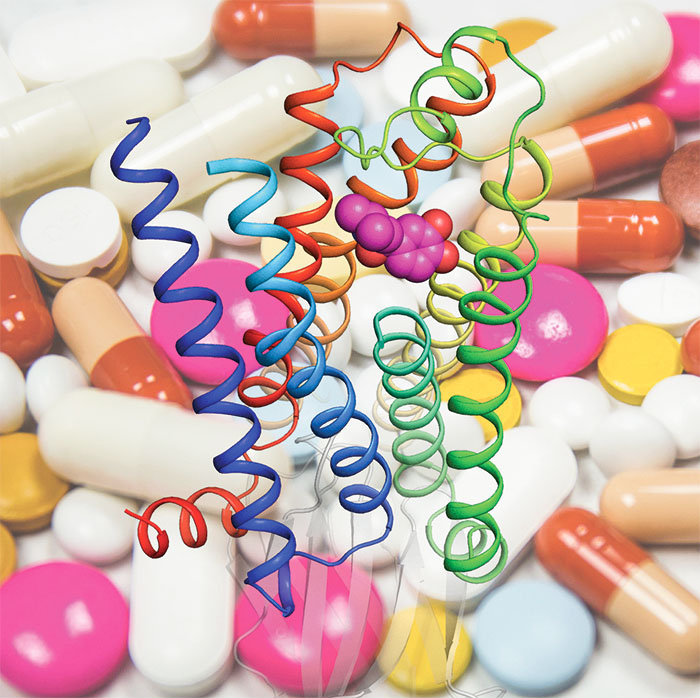
Much of the communication in cells is dependent on the presence of cell-surface receptors that detect signals in the form of messenger molecules called ligands. One large family of receptors are G protein-coupled receptors (GPCRs). This family includes a number of important drug targets, so understanding their structure and function are important. Their name derives from the fact that the receptor must couple with a G protein in order to function. One effect of this coupling of the G protein with the GPCR is increased affinity of binding of the ligand to the receptor, but how this occurs has not been well understood. Chris Tate’s group in the LMB’s Structural Studies Division, collaborating with Andrew Doré at Sosei Heptares, have now determined how coupling with a G protein leads to contraction of the binding pocket so that ligands are more tightly bound.
Over the last ten years there has been a revolution in our mechanistic understanding of the function of GPCRs. A large number of structures of receptors in different activation states have led to a detailed picture of how binding of a ligand, or agonist, such as a hormone, drives changes in the shape of the protein until it reaches an active state that will couple with a G protein. The G protein then becomes activated, which allows it to propagate signalling into the cell, as well as causing the agonist to be more tightly bound. The difficulty in obtaining multiple structures of a GPCR in both the inactive state and the activated state has been a barrier to understanding the difference between these states.
Tony Warne, in Chris’ group, used high affinity nanobodies to achieve an equivalent effect to the G protein and trap a GPCR, the beta1-adrenoceptor, in the active state while bound to one of four different agonists. For the first time, this allowed direct comparison between these structures and previously obtained structures of the same GPCR in the inactive state while bound to the same four agonists. The team found that the agonist binding pocket contracted when the beta1-adrenoceptor became activated, although the structural changes differed subtly depending on the agonist bound. Within this contracted binding pocket, they could see that there were more atomic interactions between the agonist and receptor and that the bonds between them were shorter and stronger, explaining the stronger affinity of the interaction.
Members of the GPCR family are targets for 34% of marketed small molecule drugs; drugs that treat a wide range of ailments and diseases, such as migraines, asthma, and high blood pressure. The beta1-adrenoceptor is a target for heart medication and the agonists used in this study included the drug salbutamol, which is used to treat asthma. Importantly, although the GPCR family is diverse, there is a high degree of similarity in the way in which G proteins are activated. Thus, it is likely that this new understanding of the mechanism of altered binding after G protein activation will be relevant to many other drug targets. A full understanding of the mechanisms of function of these proteins will allow design of better therapeutics that have fewer side effects.
The work was funded by the MRC and ERC.
Further references
Molecular basis for high affinity agonist binding in GPCRs. Warne, T., Edwards, PC., Doré, AS., Leslie, AGW., Tate, CG. Science [Epub ahead of print]
Chris’ group page