
 Amyotrophic lateral sclerosis (ALS) is caused by the progressive dysfunction of specific nerve cells that control muscle movement. It belongs to a group of devastating neurodegenerative diseases including Alzheimer’s, Parkinson’s, Huntington’s and prion diseases. Each disease is caused by the progressive accumulation of specific proteins in an aberrant, misfolded shape. The formation of the protein deposits is somehow toxic to nerve cells but why and how they initially form is unclear.
Amyotrophic lateral sclerosis (ALS) is caused by the progressive dysfunction of specific nerve cells that control muscle movement. It belongs to a group of devastating neurodegenerative diseases including Alzheimer’s, Parkinson’s, Huntington’s and prion diseases. Each disease is caused by the progressive accumulation of specific proteins in an aberrant, misfolded shape. The formation of the protein deposits is somehow toxic to nerve cells but why and how they initially form is unclear.
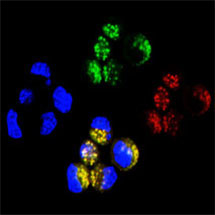
Anne Bertolotti’s group from the LMB’s Neurobiology Division has been studying the protein superoxide dismutase-1 (SOD1), which becomes misfolded in familial inherited forms of ALS. The group discovered that neuronal cells spontaneously and efficiently take up the misfolded mutant SOD1 from their environment. The internalized mutant SOD1 triggers a change in shape of the normally soluble mutant SOD1 protein, which causes its aggregation. Mutant SOD1 aggregates are then released from cells and transferred to neighbouring cells with remarkable efficiency, leading to a cycle of events that goes on indefinitely, like for prions.
Understanding the early molecular events leading to devastating neurodegenerative diseases such as ALS will lead to the design of novel and targeted therapeutic approaches. Anne Bertolotti and colleagues have already found ways to block the entry of aggregates into cells, the first step in the prion-like cycle of SOD1. The knowledge that mutant SOD1 proteins are released from cells may also facilitate the design of effective therapeutics against the mutant protein causing ALS when it is outside, rather than inside cells.
Further references:
Full article in PNAS “Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells”
Anne Bertolotti’s Group Page