
 New research, from a team of scientists in the LMB’s Structural Studies Division and the EMBL-European Bioinformatics Institute, has uncovered how our genome keeps the effects of mutations in check. The discovery, published in the journal Cell, explains how new proteins are created, helping to deliver useful insights into the evolution of the human genome. The team’s findings may also help in improving our understanding of diseases such as cancer and neurodegeneration.
New research, from a team of scientists in the LMB’s Structural Studies Division and the EMBL-European Bioinformatics Institute, has uncovered how our genome keeps the effects of mutations in check. The discovery, published in the journal Cell, explains how new proteins are created, helping to deliver useful insights into the evolution of the human genome. The team’s findings may also help in improving our understanding of diseases such as cancer and neurodegeneration.
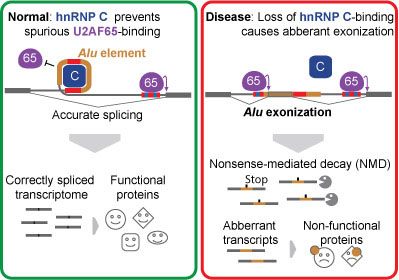
Our genes are made up of stretches of DNA called exons, which code for proteins, and introns, which do not. Prior to protein production, exons are spliced together, removing introns, by a process known as splicing. However, introns often contain short DNA segments that closely resemble the splicing signals at the ends of exons. These segments are often called ‘pseudo-exons’ because there is always a chance they will be mistaken for exons and be included in the final transcript – a potentially dangerous scenario for the organism due to the protein mutations produced. Most sequence variation among individuals occurs within the 98% of our genome that does not encode proteins. We understand very little about the function of the non-coding intron sequences and are rarely able to interpret the meaning of mutations in these regions.
Julian König and others in the LMB’s Structural Studies Division, led by Jernej Ule, together with Kathi Zarnack and long-term collaborator Nick Luscombe, from EMBL-EBI, have discovered how our genome keeps the effects of these intron mutations in check to protect the ‘transcriptome’.
One of the most common sources of intronic pseudo-exons are Alu transposable elements, of which there are about 650,000 within our genes, and these are a common cause of harmful genetic mutations. Using a technique called iCLIP, the groups were able to identify interactions between proteins and RNA within the cell at nucleotide resolution. They found that an abundant RNA-binding protein called hnRNP C dedicates a quarter of its binding sites to suppressing pseudo-exons. The hnRNP C binds to the Alu’s splicing motifs, effectively hiding them from the splicing machinery. Cells missing hnRNP C activate thousands of Alu pseudo-exons, and the proteins they produce are damaged. Many ‘disease exons’ occur because a binding site for hnRNP C has been lost, the Alu gets expressed and it creates a problem.
Because mutations involving Alu transposons are thought to be involved in diseases such as cancer and neurodegeneration, discovering how hnRNP C works may lead to new ways of mitigating the effects of potentially harmful mutations through development of treatments that target this mechanism.
The discovery also provides useful insights into how new proteins are created in the evolutionary process. Around 5% of alternative exons in the human transcriptome originate from bona fide exonization of Alus and some of these are thought to be unique to the human genome. Therefore, although intronic Alus generally pose a massive threat to transcriptome integrity, they also offer opportunities for evolutionary innovation. The repressive function of hnRNP C prevents the damaging effects of immediate Alu exonization, and thereby enables mutations to gradually create Alu-derived exons. This represents an elegant molecular mechanism that could mediate incremental evolution of new cellular functions.
The work was funded by the MRC LMB, the European Research Council, an EMBL EIPOD fellowship and a Long-term Human Frontiers Science Program fellowship.
Further references:
Cell paper
Jernej Ule’s group page – now at UCL
Nick Luscombe’s group page