
The DNA in cells is constantly damaged by both internal activities of the cell and by external factors such as ionising radiation. In order to function correctly, this damage must be repaired, or if it cannot be repaired, the cell must be killed to prevent development of diseases such as cancer. The large protein kinase, ataxia-telangiectasia mutated (ATM), is a vital component of the cell’s DNA repair machinery.Research led by Roger William’s group in the LMB’s PNAC Division, in collaboration with a team at AstraZeneca’s IMED Biotech Unit led by Chris Phillips, has for the first time revealed the near-atomic structure of ATM.
A cell may experience up to one million lesions in its DNA in a single day. ATM is responsible for a cell’s responses to double-strand DNA breaks. These can be especially damaging, since they can give rise to gene rearrangements. An early event brought about by these breaks is the recruitment of ATM to the site of the damage, where it becomes activated and triggers a wide range of responses to the damage, including arresting the cell cycle by phosphorylating the checkpoint kinase CHK2 and, under some conditions, inducing apoptosis by phosphorylating p53 and preventing its degradation.
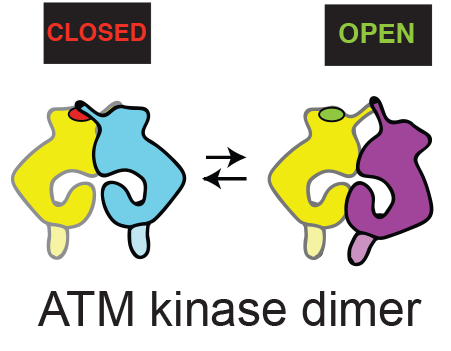
Using cryo-EM, Domagoj Baretić, in Roger’s group revealed that ATM is in a dynamic equilibrium between inactive and active dimers. More than half of the ATM structure consists of an N-terminal helical solenoid. Analysis by Balaji Santhanam at the LMB showed that, despite some structural similarity with other kinases, the N-solenoid has a largely unique sequence. This solenoid binds regulatory proteins and might alter ATM activity by controlling the abundance of active and inactive conformations of the dimer. Although earlier non-structural work with ATM had proposed that ATM is activated by a dimer-to-monomer transition, no monomers were detected using cryo-EM. Furthermore, analysis of ATM in solution by Chris Johnson, head of the LMB Biophysics facility, was consistent with the enzyme being a dimer. Using p53 produced by Tomas Kouba in Alan Fersht’s group, it was shown that this dimeric ATM is an active kinase. Hannah Pollard, David Fisher, and Caroline Truman at AstraZeneca initially cloned, expressed and characterised the ATM.
Mutations that prevent ATM function in adult brains lead to an impaired ability to correctly maintain the neuronal population, with dysfunction especially pronounced in the cerebellum, giving rise to the characteristic ataxia for which ATM was named. However, the consequence of ATM loss has many other profound effects on an individual, including development of T-cell lymphomas. Revealing the structure of ATM is providing insights into how this important enzyme functions and may aid the production of specific inhibitors of ATM that could make anti-cancer therapeutics more effective.
This project is funded through a research collaboration between AstraZeneca UK Limited and the Medical Research Council (LMB/AstraZeneca Blue Skies Initiative).
Further references:
Paper in Science Advances
Roger’s group page
LMB News: LMB and AstraZeneca announce plan to launch joint research fund