
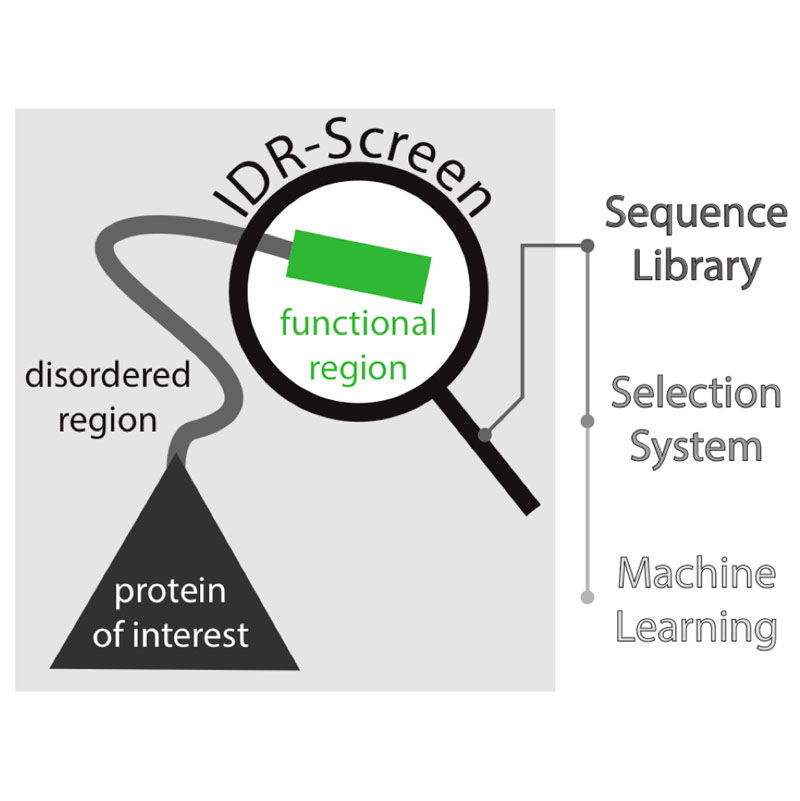
Work from Madan Babu’s group in the LMB’s Structural Studies Division, spearheaded by Charles Ravarani and in collaboration with Alexandre Erkine’s group at Butler University, has for the first time harnessed next generation sequencing and machine learning to develop a high throughput screen to uncover disordered regions of proteins that are functional within cells.
Proteins, the molecular machines of the cell, are formed from chains of amino acids. Understanding how the amino acid sequence of a protein contributes to its function has long been a problem of interest. Whilst some amino acid sequences fold into defined three-dimensional structures, many proteins have intrinsically disordered regions (IDRs). For decades these regions were ignored as they were thought to be non-functional. However, it is becoming clear that they are implicated in a number of human diseases, such as cancer and neurodegeneration, and often contribute to the protein’s function by mediating transient interactions with other molecules in the cell. Importantly, a major limitation to studying disordered proteins has been the lack of large-scale approaches that allow for the discovery of functional disordered regions in biologically relevant contexts.
One type of protein that relies on a disordered region to perform its function is transcription factors – the transactivation domain of transcription factors is typically unstructured and can recruit the transcription machinery to promoters, leading to transcription of genes. Although transactivation domains have been extensively studied, the molecular rules of what makes an unstructured sequence function as a transactivation domain remain poorly understood. To better understand the disordered regions in transactivation domains and crucially to provide a tool with which IDRs can be studied more broadly, the team of scientists developed an IDR-Screen which allows the discovery and analysis of disordered regions that are functional within cells.
First a library of cells containing a large number of different disordered sequences is created, and the cells are screened to identify those that contain a functional sequence. The team used a specific transcription factor in yeast, replacing its transactivation domain with a library of sequences which code for disordered segments. If the disordered segment is non-functional, the transcription factor cannot initiate transcription of downstream genes and the yeast cell dies. Cells that survived must therefore contain disordered regions that can function as transactivation domains. Next generation sequencing is then used to decode the functional sequences that are present in the surviving cells and machine learning is employed to understand what features give these sequences their function. In the case of transactivation domains, the IDR-Screen revealed that functional domains lack positively charged amino acid residues but have short sequence patterns of negatively charged and aromatic residues.
Computational approaches have estimated that there could be up to a million functional IDRs in the human proteome. However only a small fraction of these have been characterised so far. This new research and IDR-Screen developed by Madan’s group represents an important advance as this technology allows for the discovery and characterisation of biologically and functionally relevant disordered regions from any genome as well as enhancing our understanding of what features make such sequences functional. The approach can be readily expanded to study other functions, beyond transactivation domains, that are mediated by disordered regions such as protein degradation, and can be used to understand the effect of mutations in disordered regions that are seen in cancer genomes and in the normal human population. The better we understand these disordered segments, the more we are in a position to understand the molecular basis of diseases that they cause, as well as design drugs to target them.
This work was funded by the MRC, the ERC, the AFR Scholarship from the Luxembourg National Research Fund, a Marie Curie Fellowship, the National Science Foundation, the Lister Institute Research Prize and the Holcomb Award and Innovation Grant from Butler University.
Further references:
Paper in Molecular Systems Biology
Madan’s group page
Alexandre Erkine’s group page
A million peptide motifs for the molecular biologist
What is the dark proteome
Illuminating the dark proteome