
Mass Spectrometry

Mass spectrometry based proteomic experiments have become an important tool for studying biological systems. We implement state-of-the-art mass spectrometric techniques in order to provide scientists at the LMB with the capabilities to identify, characterise and quantify proteins present in a particular network, pathway, organelle, cell or tissue.
We principally employ electrospray ionisation mass spectrometry for both qualitative and quantitative characterisation of proteins. The basic approach for protein analysis is ‘bottom-up’ proteomics, where proteins are initially enzymatically digested with proteases producing a peptide pool representative of the original proteins.
Qualitative protein identifications and the identification of post-translational modifications are performed using nano-scale capillary LC gradient separations coupled to Orbitrap mass spectrometers (ThermoFisher LTQ XL, Velos, QE, QE+ and QE-HFX).
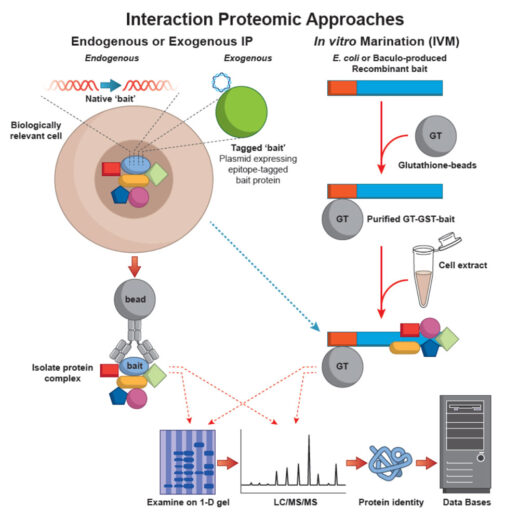
We utilise three techniques for mass spectrometric based relative protein quantification. Differential protein expression can be performed using either an isotope labelling technology (ThermoScientific TMT reagents), Stable Isotope Labelling by Amino acids in cell Culture (SILAC), which relies on metabolic incorporation of the quantitative label or a completely “label free” approach.
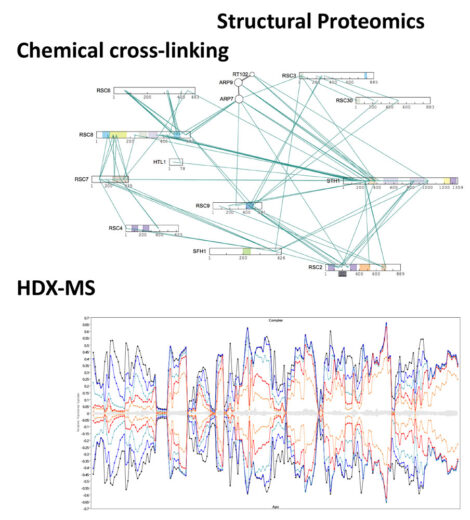
The development of new mass spectrometric-based methods is essential in order to ensure that the LMB remains at the forefront of biological research. The group undertakes independent research to create new mass spectrometric technologies and we collaborate with LMB groups and external laboratories to develop and apply these bespoke methods to answer important biological questions. The major theme of research within the facility involves the development of Structural Proteomic methods for characterising protein/protein and protein/nucleotide interactions. Major efforts involve the use of chemical cross-linking to investigate both transient and stable interactions within protein complexes, and hydrogen deuterium exchange-MS (HDX-MS) for the elucidation of protein-protein, protein-nucleotide and protein-ligand interactions.
Selected Papers
Shakeel S, Rajendra E, Alcón P, O’Reilly F, Chorev DS, Maslen S, Degliesposti G, Russo CJ, He S, Hill CH, Skehel JM, Scheres SHW, Patel KJ, Rappsilber J, Robinson CV, Passmore LA. (2019)
Structure of the Fanconi anaemia monoubiquitin ligase complex.
Nature. 575(7781):234-237.
Das S, Malaby AW, Nawrotek A, Zhang W, Zeghouf M, Maslen S, Skehel M, Chakravarthy S, Irving TC, Bilsel O, Cherfils J, Lambright DG. (2019)
Structural Organization and Dynamics of Homodimeric Cytohesin Family Arf GTPase Exchange Factors in Solution and on Membranes.
Structure. pii: S0969-2126(19)30313-2. doi: 10.1016/j.str.2019.09.007. [Epub ahead of print]
Famelis N, Rivera-Calzada A, Degliesposti G, Wingender M, Mietrach N, Skehel JM, Fernandez-Leiro R, Böttcher B, Schlosser A, Llorca O, Geibel S. (2019)
Architecture of the mycobacterial type VII secretion system.
Nature. doi: 10.1038/s41586-019-1633-1. [Epub ahead of print]
Williams TD, Peak-Chew SY, Paschke P, Kay RR. (2019)
Akt and SGK protein kinases are required for efficient feeding by macropinocytosis.
J Cell Sci. 132(2).
Gillingham AK, Bertram J, Begum F, Munro S. (2019)
In vivo identification of GTPase interactors by mitochondrial relocalization and proximity biotinylation.
Elife. 11;8. pii: e45916.
Gladkova C, Maslen SL, Skehel JM, Komander D. (2018)
Mechanism of parkin activation by PINK1.
Nature. 559(7714):410-414.