


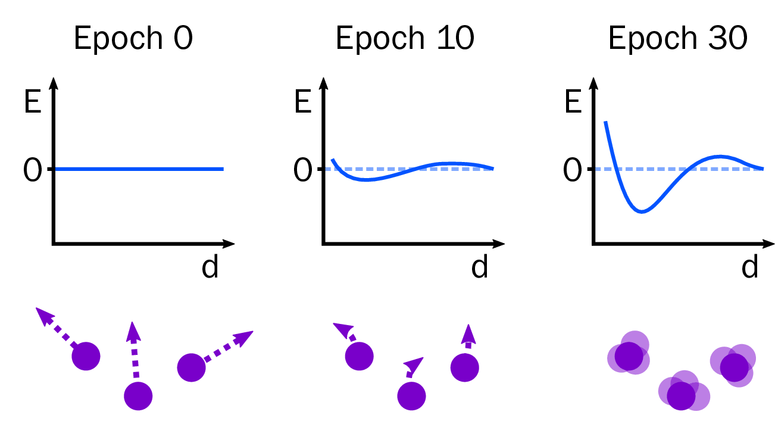
Molecular dynamics has shown success in obtaining biological insights by providing mechanistic interpretations of experimental data. However, the force fields used to describe how the atoms interact are biased towards keeping folded proteins folded and fail when applied to disordered proteins or protein aggregation. The idea of using the technique of automatic differentiation, most commonly used to train neural networks, to improve force fields is called differentiable molecular simulation (DMS). It overcomes the limitations of other methods by allowing multiple parameters to be tuned at once.
We aim to use DMS to improve all-atom force fields so they can work across a variety of protein systems, allowing us to answer questions of biological importance involving disordered proteins and disease-associated protein aggregation. We also aim to use DMS to develop new force fields at different scales, such as implicit solvent or coarse-grained models, allowing larger systems to be simulated. The next few decades will see compute resources increase to the point where simulations at biologically relevant length and time scales become routine. It is crucial that we have accurate physical models available to take advantage of this and to probe the molecular basis of life.
Selected Papers
- Greener JG and Jones DT (2021)
Differentiable molecular simulation can learn all the parameters in a coarse-grained force field for proteins
PLoS ONE 16(9): e0256990 - Greener JG, Kandathil SM and Jones DT (2019)
Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints
Nature Communications 10: 3977 - Greener JG, Moffat L and Jones DT (2018)
Design of metalloproteins and novel protein folds using variational autoencoders
Scientific Reports 8: 16189 - Greener JG, Filippis I and Sternberg MJE (2017)
Predicting protein dynamics and allostery using multi-protein atomic distance constraints
Structure 25: 546-558